Orbitais
Espectroscopia: ciência que nos permite saber informações sobre as estruturas microscopicas. Estuda a interacção do espectro electromagnético com a matéria.
- Nivel atómico - raio gama.
Mecânica quântica: mecânica das particulas atómicas.
Natureza ondulatória da luz: 2 ondas interferem entre si (dado a sua propriedade sinusal).
- Em fase: λ mantêm-se, ν duplica.
- Em fase oposta: anulam-se.
- Espectro: continuo / descontinuo.
- Propriadades fisicas:
- macroscopica (continua)
- microscopica (não continua)
Principio da incerteza de Heisenberg: não podemos determinar com precisão a localização de particulas microscopicas.
- Utilizamos uma função que exprime a probabilidade da localização da particula.
Equação de Schrodinger:
- HΨ=EΨ
- Ψ: função de onda.
- E: energia.
- H: hamiltoniano (dá os niveis de energia permitidos para esse sistema).
- Soluções: números, vectores, funções, conjuntos de funções.
- Após a Equação de Schrodinger: E = R / n^2
Números quanticos:
- n: quantifica E.
- l: quantifica o momento ângular de orbital (0 ≤ l ≤ n-1).
- s: quantifica o momento ângular de spin.
- ml: quantifica a projecção dos momentos angulares (-l ≤ ml ≤ l).
Equação de Schrodinger: um conjunto de funções que descreve a interação do sistema.
Superficies nodais: zonas onde a probabilidade de encontrar o electrão é de 0%.
- n = 1 - 0
- n = 2 - 1
- n = 3 - 2
Estrutura molecular
Reactividade: comportamento na presença de outras substâncias.
Diamagnético: não reage a um campo magnético, electrões em número par e emparelhados, momento magnético = 0.
Paramagnético: reage a um campo magnético, electrões desemparelhados cujo número determina o momento magnético.
- Ex. μ = 1.0 MB (magnetões de Broun).
Tipos de ligação
- Orbitais moleculares: descrever.
- Ligação covalente: muitos atomos, é mais simples.
- Modelo de repulsão de pares electrónicos: a ligação é dada entre 2 electrões. Permitiu reconhecer que existem pares de electrões compartilhados e não compartilhados.
- Teoria da ligação de valencia: existem orbitais atómicas. Aproximando 2 atomos há uma distância em que existe uma interacção favorável - 2 orbitais sobrepõem-se e estabelecem uma ligação mais favorável.

- Tipo sigma σ: distribuição da densidade electronica em torno de um eixo. Simetria esferica - oval / cilindrica. Sobreposição topo a topo.
- Tipo π: sobreposição de moleculas lado a lado, surge um plano nodal no eixo nuclear. Densidade electrónica sobre um plano.
- Orbital atómica hibrida: combinações lineares de outras orbitais. As orbitais são funções, podemos somar funções.
- Carbono: esperava-se 2 ligações mas forma 4. Os 2 electrões da 2s passam para 2p, logo são 4 electrões desemparelhados. Fusão das orbitais s e das p (todas com a mesma energia), formam ângulos de 90º - tetraedrico. sp3: 2s + 3p.
- Carbono planos: um electrão fica na 2pz e forma ligações do tipo π. sp2: 2s + 2px + 2py.
- Hibridação ap: s + p, ângulo 180º.
- sp3d: bipiramidal trigonal, 5 ligações.
- sp3d2: octaedrica, 6 ligações.
- Benzeno: densidade electronica sobre e sob a molecula, electrões das ligações π na orbital 2sp. Ligações C-C e C-H em orbitais sp2.
Energia de ligação (E.L.)
- Maiores atomos → maior afastamento → maior comprimento de ligação → menor energia de ligação.
- 1s (H) com 2sp3 (F) < comprimento de ligação, > E.L., > sobreposição
- 1s (H) com 3sp3 (CL) > comprimento de ligação, < E.L., <sobreposição
- sp3d: bipiramidal trigonal, 1.s + 3.p + 1.dyz.
- orbitais d: dxy, dxz, dyz, dx2y2, dz2.
- sp3d3: octaedrica, 1.s, 3.p, dxy, dxz.
Teoria das orbitais moleculares

- Após a formação da molécula → orbitais moleculares.
- Eq. Schrodinger para molécula → funções orbitais moleculares.
- Se ocuparem o espaço (zl), os electrões favorecem a ligação.
- Se ocuparem o resto desfavorecem - orbitais anti-ligantes, maior energia.
- Zona ligante: orbital molecular ligante.
Ordem de ligação, OL
- OL = (nº de electrões ligantes - nº de electrões não ligantes) / 2
- ex. H2+ OL=1/2
- ex.He2 OL=0
Orbitais p
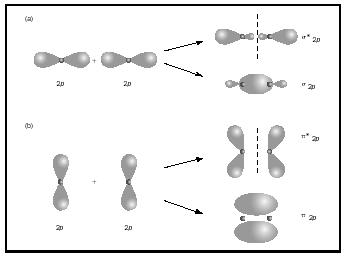
- Topo a topo: σ2p, orbital molecular ligante σ.
- Lateral: 2πp, orbital molecular ligante π.
Elementos essenciais à vida
- Macroelementos: grandes quantidades.
- H, C, N, O, Ca, P, S
- iónicos: N, K, Cl
- Elementos vestigiais: pequenas quantidades.
- Mg - relacionado com a formação de ATP.
- V - essencial a bacterias.
- I -hormonas tiroideias.
- Cr, Mn, Fe, Co, Ni, Cu, Zn, Se, Mo
- Essencial: sem ele não existia vida.
- Biodisponibilidade: quantidade do elemento diferente da presente antes da existencia de vida.
- Distribuição e espaciação: como está distribuidas e as diferentes moleculas que constitui.
Metaloproteinas
ex. citocromos, hemaglobina, mioglobina, ferrotina, transferina...
Alterações da concentração de Fe:
- Anemia ferropenica: carência.
- Ferropexia: retenção.
Os metais são normalmente o centro activo das metaloproteinas.
- Mioglobina: o centro metálico (Fe) controla a entrada e saida de O2.
- Hemoglobina (tetramero): transporta 4 O2.
- Monomeros: α1, α2, β1, β2.
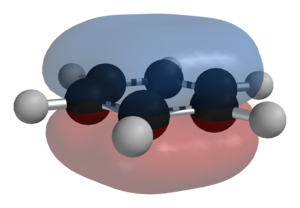
Cada Fe2+ liga-se a uma porfirina ligada a uma cadeia proteica que impede a oxidação do Fe. - Porfirina: espaço central com 4 atomos de N. Os 2 electrões livres estabelecem ligação com o Fe2+, que forma 4 ligações com 4 N e ainda tem tendencia a formar 2 ligações com o O2.
- O2: ligação reversivel. CO: ligação irreversivel.
- Sem O2: a ligação é mais facil à medida que se ligam e o fenomeno é cooperativo.
- Vitamina B12: metaloproteina de centro activo com Co2+. Cataliza reacções de transferencia de H, grupos carbonilo ou carbonados.
- Citocromos: metaloproteina que permite a transferencia de electrões (oscila entre Fe3+ e Fe2+).
- Purina: poro que permite a passagem. ex. aquapurina.
A luz dos iões metalicos de transição resulta dos electrões das orbitais d (incolor quando não há transições electronicas).
Separação
- Homolítica
- Heterolitica
Coordenação do oxigénio molecular: O2 liga-se ao fe e há uma ligeira contracção que se deve ao movimento de spin do Fe2+.
Ácido de Lewis: substâncias capazes de aceitar densidade electronica.
Base de Lewis: substâncias capazes de ceder densidade electronica.
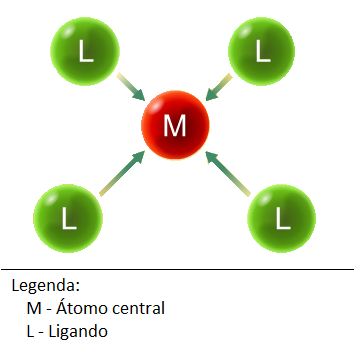
Complexo de metais de transição:
Geometrias

Complexo de metais de transição:
- Ião central: ácido de Lewis (aceitador de electrões).
- Ligando: base de Lewis (dador de electrões; tipo e densidade).
- Modelo de ligação: esquema dador-aceitador / ligações.
Composto de coordenação: compostos onde existem ligações covalentes, também designados por complexo de metais de transição.
 |
| Ligações coordenadas (covalentes fortes). |
Ião metálico de transição - são todos ácidos de Lewis.
Geometrias
- CN → nº de ligações coordenadas é caracteristico do ião e dos ligandos.
- CN → indica o nº máximo de ligações que o ião estabelece.
- CN = s (adquire a geometria mais adequada)
Isomeros
- Cis (lateral), trans (transversal), posição equatorial e axial.
Ligandos:
- Bidentado: pode ligar-se a dois locais. Ex. etileno diamina.
- Monodentado: pode ligar-se a um local. Ex. água, amoniaco.
- Ambidentado: pode ligar-se a 2 iões metálicos (como é linear têm que ser diferentes). Ex. tiocinato.
- Tetradentados: pode estabelecer 4 ligações. Ex. porfirina.
→ Depende dos atomos capazes de dar densidade electrónica. Varia o nº de ligações de acordo com o nº de átomos que cedem densidade electrónica. Os iões metálicos de transição têm muita tendencia a formar compostos de coordenação. A estabilidade da molécula depende da força das ligações.
Estabilidade de uma substância: relaciona-se com a energia de ligação, várias espécies em conjunto têm maior energia do que separados.
- Avaliada por Constante de Formação (ex. 10^15 → sentido de formação de produtos)
- Constante global: de todos os ligandos.
- Constante parcial: de cada ligando.
→ Constantes de estabilidade de complexos / Constante de equilibrio de complexidade
A densidade dos ligandos é importante para a estabilidade da ligação. É influenciada pelo nº de moleculas ligandos: constante é maior, menor o nº de ligandos → se a formação provier de um só ligando é mais estável.
Propriedade Quelante dos Ligandos: para terem, os ligandos têm que ser polidentados. Quanto menos moléculas estabelecerem ligações, maior a probabilidade quelante desse ligando.
- Quanto mais estável → mais se desenvolve no sentido dos produtos → menos iões livres → produto muito estável.
- Diferentes ligandos → diferentes complexos → diferentes estabilidades.
Ligandos Polidentados
 |
| Bidentado: etilendiamina |
 |
| Tridentado: terpiridina |
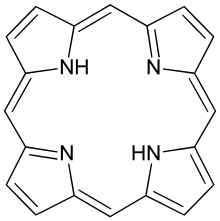 |
| Tetradentado: porfirina |
 |
| Hexadentado: EDTA |
Equilibrio de complexação
- Ligando monodentado neutro: não interfere na carga do ião metálico.
- A formação de complexos ocorre em reacções de equilibrio, caracteristicas pelas suas constantes (constante >1 → reacção muito extensa).
- β3 = k1 . k2 . k3 (quanto maior o valor de k, mais deslocado é o equilbrio no sentido dos produtos).
- Se substituirmos dois ligandos monodentados por um ligando bidentado, a Keq e a estabilidade é diferente.
- O Keq é diferente se ligarmos 2 ligandos monodentados e 1 ligando bidentado devido ao efeito quelato.
Reacções de transferencia protónica e electrónica
- A variação do estado de oxidação depende dos ligados e do ião metálico.
- Tem que haver uma entidade que ceda electrões e outra que receba.
- Parametros: dizem a tendencia da substancia em se oxidada-reduzida (há um termo de comparação: H2).
- Capacidade de redução-oxidação: varia com a molecula onde o ião está inserido e com o próprio ião metálico.
Diagrama de especiação: saber as substâncias que existem num meio de pH x.
Modelos de ligação
- Metais de transição: caracteristicas estruturais → caracteristicas espectometricas especificas (coloridos).
As suas configurações electrónicas dos complexos de metais de transição são afectadas pelos ligandos ligados ao ião central. - Teorias de ligação
- Ligação de valencia: pode ser usada desde que não nos esqueçamos que a densidade electrónica é cedida pelos ligandos (o ião central recebe densidade electrónica dos ligandos, permanecendo os seus electrões de valencia - 3d - sem intervir nas ligações).
- Teoria de ligação de valencia
- Ligações formadas através da sobreposição das orbitais vazias do metal com as orbitais dos ligandos.
- Electrões de valencia (3d do metal) não fazem ligação (só é à "custa" dos electrões dos ligandos).
- Teoria do campo de cristal
- Todos os ligandos possuem carga negativa e influenciam o ião central.
- A carga negativa repele os electrões das orbitais d (a aproximação dos ligandos leva a uma alteração de energia e forma das orbitais d).
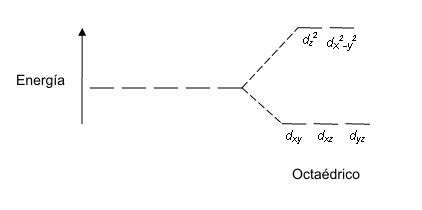 |
| À esquerda: orbital d degenerado (ião metálico isolado) À direita: orbitais d não degeneradas, valor entre elas é Δ. |
- As orbitais com menor energia são menos influenciadas pelos ligandos.
- O electrão ocupa uma das orbitais com menor energia.
- Δ : diferença de energia das orbitais d não degeneradas num campo de cristal.
- A energia e divisão das orbitais d não degeneradas depende da geometria do complexo.
- Se a energia das orbitais é toda igual, quando é irradiado, o electrão não se excita.
- Cor: sempre associada a transições electronicas d-d.
- A energia do campo de ligandos depende do ião metálico e dos ligandos.
- O campo de ligandos faz com que as orbitais deixem de estar degeneradas.
spin baixo: s=1 → 2 electrões desemparelhados
spin alto: s=2 → 4 electrões desemparelhados
momentos magnéticos diferentes
→ Os electrões vão adquirir a configuração electrónica mais favorável em termos energéticos.
Nota: eg = dz2 e dx2y2 ; t2g = dxy, dxz, dyz.
Δo - variação de energia de campo ligando correspondente a uma geometria octaédrica (região de luz visivel).
Série electroquimica de ligandos: agrupa os ligandos de acordo com as cores que as soluções apresentam.
- ΔE = hν = hc / λ
- Ligandos que dão origem a campos fortes têm ligações tipo π → excelentes dadores de σ e excelentes receptores de π.
- Ligação π → ligação mais forte, ΔE maior, compostos de spin baixo.
- O spin é muito importante e influencia o comportamento das substâncias.
Geometria octaedrica: só é perfeita se todos os ligandos forem iguais (mesma distância do centro metálico).
CN = 4 → geometria tetraédrica ou quadrado plano (xy).
Geometria tetraedrica: a influencia dos ligandos vai ser maior sobre as bissectrizes dos planos (orbitias dxy, dxz, dyx). Ocorre o oposto na geometria octaedrica.
Geometria em quadrado plano: ião cenral e ligandos no mesmo plano (xy). É como a geometria octaedrica menos os dois ligandos do eixo do z.
- dz2 baixa a sua energia dado os ligandos não influenciarem segundo o eixo dos z's.
- dx2y2 vai ser a mais energética de todas.
- dxy vai sentir maior influencia dos ligandos.
Energia da orbital → energia da cor absorvida → ΔE = hν
- Composto de coordenação com electrões de transição têm propriedades diferentes de electrões representativos (electrões nas orbitais d).
- Os electrões das d não são ligantes mas sofrem influencia dos ligandos.
- Sem a influencia dos ligandos, as orbitais têm todas a mesma energia.
Propriedades espectroscópicas a nivel da cor
- Cor: transição d-d; branco: não são transições d-d, são outras.
- As propriedades magnéticas também são importantes.
- Spin baixo: spin oposto, campos magneticos anulam-se.
- Δ: diferença de energia do campo de ligandos. P: energia de emparelhamento.
- Δ < P: electrões vão querer nivel de energia menor, não se emparelham, ocupam orbitais mais energeticas.
- Δ > P: electrões querem ocupar a mesma orbital, emparelhar e gastar energia com o spin.
- Nota: Δ pequeno (ligando fraco) adsorvem λ grandes, cor de λ pequenos.
- Orbitais d preenchidas → não há transições → não apresenta cor → pode ter cor devido a outros ligandos / iões na solução → não podemos calcular ΔE pela cor, mas não significa ΔE=0.
- Spin: não interfere com a estabilidade (constante de formação), não interfere com as reacções acido-base de Lewis.
- Estabilidade: E conjunto < E elementos separados, a molécula é estável a partir do momento que não se decompõe.
- Keq: extenção da reacção.
- K = (produto da | | produtos) / (produto da | | reagentes)
- K=1 não tende
- K>1 mais produto
- K<mais reagente
- Ligando forte ou fraco origina campo magnético forte ou fraco.
Teoria do campo ligando
- Os ligandos são entidades capazes de dar / aceitar densidade electrónica.
- Ligandos de campo fraco: excelentes dadores de σ.
- Ligandos de campo forte: excelentes dadores de σ e aceitadores de π.
- Teoria das orbitais moleculares: os electrões d estão em orbitais moleculares não ligantes (responsáveis pelas propriedades), a energia das orbitais ligantes do complexo é inferior à energia das orbitais que lhe deram origem (com a formação do complexo há um ganho de energia).
Elementos essenciais
- Patologias: carencias e excesso.
- Diabetes melitus tipo I: admnistração de iões metalicos (zinco e vanadium) levam a uma diminuição a nivel de glicose no sangue (a insulina não pode ser administrada oralmente - não resiste ao pH do estomago).
- Metodos de diagnóstico - ressonancia magnética: observar o momento magnético em termos de protões da água. Agentes de contraste - iões metálicos que melhoram o sinal dos protões (Godolinum e Disprósio em complexo).
Alcanos
- Ligações C-C e C-H.
- Lineares ou ramificadas.
Carbono
- 4 electrões de valencia.
- Hibridação sp3 (como têm muito componente p, estas orbitais são mais estendidas para fora do que concentradas) → sobrepõem-se às s do H formando ligações fortes e sigma (cilindrica que permite rotação).
- Geometria tetraedrica: 109.5º (afastamento máximo)
Etano
- Ligação sigma C-C, sp3 que se sobrepõe às s do H.
- Geometria em zig-zag.
Propriedades fisicas
- Serie homologa de compostos: metileno (CH2).
- Temperatura: 25º, 1 atm.
C1 a C4: gases; C5 a C17: liquidos; >C18: sólidos. - Ponto de ebulição: aumenta linearmente com o número de carbonos.
- Ponto de fusão: não respeita o nº de carbonos. Há alternancia entre pares e impares, apesar de cada um aumentar linearmente.
- Densidade: <1, são mais leves que a água.
- Solubilidade: pouco polares e incapazes de ligações com H, logo pouco soluveis em água. Soluveis uns nos outros e em solventes de baixa polaridade.
- Electronegatividade: capacidade de atrair electrões → H e C muito semelhantes → ligação no metano apolar → é uma partilha muito simétrica → sigma pura (covalente).
→ Polaridade negativa: atrai para si electrões.
→ Polaridade positiva: deficiencia electrónica. - Conformação: rotações em torno de uma ligação sem partir (a energia vai alterando ligeiramente).
Cicloalcanos
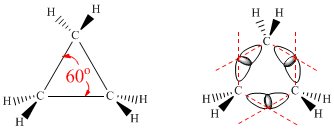 |
| Ciclopropano: 60º ≠ 109.5º, comprime e dobra orbitais. Tensão angular logo muito instavel. |
 |
| Propano: planar, H eclipsados, instabilidade. |
- Ciclobutano: ângulo interno 88º, dobra as orbitais, tensão angular. Estrutura ligeiramente dobrada, H não eclipsado.
- Ciclopentano: ângulo interno 108º (próximo de 109.5º) - estável. Conformação ligeiramente dobrada para evitar eclipse.
- Ciclohexano.
 |
| Conformação em cadeira. A vermelho axiais (perpendiculares ao plano), a azul equatoriais (horizontais ao plano). Equatorial passa a axial e vice-versa. |
 |
| C-C: sp3 flexivel. |
Conformação em cadeira
- Ângulo: 109.5º - não há tensão angular.
- Grupos desencontrados - não há tensão torcional.
- Máximo distância - livre de repulsões de Van der Waals,
Conformação em barco
- Ângulo: 109.5º - não há tensão ângular.
- Hidrogénios eclipsados - há tensão torncional.
- H nos C1 e C4 muito próximos - há tensão de Van der Waals.
Análise conformacional
- Estudo da variação da energia de acordo com a rotação.
- Quando o ciclohexano tem substitutos de H, a conformação mais estável é a que esse grupo está em posição equatorial.
- Equatorial: mais estável, menos energia, sem repulsões de Van der Waals.
- Axial: fica aglomerado a H axiais, há repulsões.
Estereoquimica
Química que lida com o raio espacial da molécula.
- Isomerismo: compostos diferentes com a mesma forma molecular.
- Isomerismo constitucional: isomeros em que os atomos se ligam numa ordem diferente.
- Estereoisomeros: isomeros constitucionais, diferentes arranjos dos atomos no espaço.
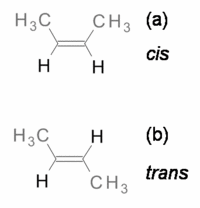 |
| Cis: grupos do mesmo lado. Trans: grupos de lados opostos. |
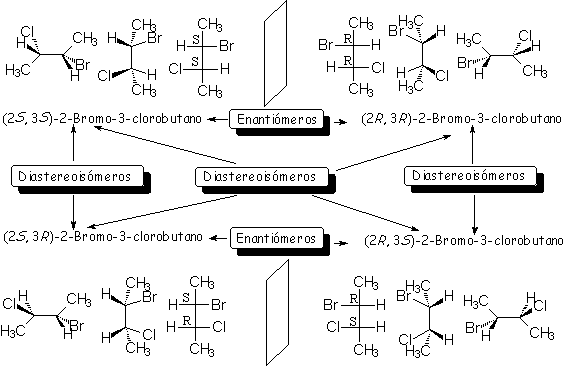 |
| Enantiomeros: estereoisomeros que são imagens no espelho. Diastereoisomeros: esteroisomeros que não são imagens no espelho. |
- Molecula Quiral: não sobreponivel à sua imagem no espelho, enantiomero.
- Esterocentro / centro estereogenico / carbono quiral: um atomo ligado a grupos de modo a que uma troca de quaisquer dois grupos resultará num estereoisomero.
- Para mudar a configuração é necessaria a quebra de ligações.
- É possivel ter um par de enantiomeros para todas as moleculas que contenham um atomo tetraedrico ligado a 4 grupos diferentes.
- C quiral pode ser sp3 ou sp2 → sp3 nas moleculas biologicamente activas.
- Se tiver dois grupos iguais não possui esterocentro.
- Plano de simetria: plano imaginario que bissecta uma molécula.
- Simetria: aquiral → não tem enantiomeros.
- Não simetrica: quiral → pode ter enantiomeros.
Nomenclatura de Enantiomeros

- Número atomico dos atomos directamente ligados ao esterocentro: maior numero atomico, maior prioridade → a, b, c, d.
- Dois carbonos: prioridade ao que estiver ligado a maiores numeros atomicos.
- Rodar a molecula de forma a que fique em sequencia de prioridades (o menos prioritario mais afastado).
- Enantiomeros: um é R outro é S.
- Ligações duplas e triplas são assinaladas como se ambos os atomos fossem duplicados ou triplicados.
Propriedades de Enantiomeros
- Mesmas propriedades fisicas: ponto de fusão e ebulição, indice de refracção, solubilidade, velocidade de reacção com reagentes normais.
- Diferenças: rotação optica (luz planar polarizada), interacção com moleculas quirais → diferentes velocidades de reacção e solubilidade.\
Luz planar polarizada: só pode passar planarmente numa unica direcção.
- Enantiomeros: quando atravessado, o plano de polarização roda.
- Rodam com a mesma grandeza mas em sentidos opostos.
Polarimetro
Mede a capacidade do composto (enantiomero) rodar a luz planar polarizada.
- Fonte de luz, polarizador (cristal → luz passa num só plano), tubo com a amostra em solução, analisador (=polarizador), escala para medir o nº graus de rotação da luz.
- Branco: não roda, composto não opticamente activo.
- Preto: rodou tudo, a luz não passa.
- Cor: luz não rodada, substância opticamente activa, molecula quiral.
- Ajustar até ver tudo branco.
- Dextrogénio: rodar para a direita.
- Levogénio: rodar para a esquerda.
→ fenomeno fisico experimental, não se relaciona com R e S. - Rotação especifica: nº de graus que o plano de polimerização é rodado (depende do nº de moleculas quirais que a luz encontra → concentração do enantiomero e comprimento do tubo → mais moleculas roda mais).
- Identificar o composto: [α] = α / (c.l)
- c: concentração ou densidade da solução (g/ml).
- l: comprimento do tubo (dm).
- α: rotação observada.
- Depende da concentração, temperatura e comprimento de onda da luz.
- [α] positivo: dextrogenio.
- Enantiomeros: [α] com valor absoluto igual mas sinal oposto.
- Rotação total: a observada, soma das rotações individuais.
Origem da actividade optica
- Molécula aquiral: a luz planar polarizada bate numa molecula e depois noutra com orientação oposta, não ha rotação.
- Molécula quiral: a luz passa e vai rodar.
- Mistura de 1:1 de (R) e (S) é equimolar → a luz passa, há cancelamento de rotação.
- Mistura racérica / racematos: compostos opticament activos, mas há cancelamento da rotação no tubo.
- Pureza enanteomerica: 100% → 1 enantiomero apenas.
% pureza enanteomerica = 100. (moles enantiomero1 . moles enantiomero2) / (moles de 1+2) - Pureza optica: podemos ver a pureza enanteomerica.
% pureza optica = 100. (rotação especifica observada) / (rotação especifica do enantiomero puro). - Pureza optica 50%: mistura racerica, 50% cancela-se uma ao outro.
Sintese de Enantiomeros
- Reacção de adição de hidrogenio: adição na mesma fase (syn) - do mesmo lado (R), do outro lado (S).
- Enzima esteroselectiva: dentro de dois enantiomeros (S) e (R) apenas escolhe um, o (S). O ácido piruvico fixa a enzima em cima e só pode ligar de um lado - reacção esteroselectiva.
- Ex. enzima acido lactivo desidroferase.
- Aparelho diafractório: cristalografia de raio X → ver a disposição dos grupos.
- Enantiomeros: difere na rotação, um carbono estereogenico.
- Diasteromeros: difere nas propriedades, mais do que um carbono estereogenico.
- Compostos Meso: molecula aquiral ocm plano de simetria, tem dois carbonos quirais, opticamente inactiva, imagem no espelho e composto sobreponiveis. O plano de simetria dissecta a molécula em duas metades iguais.
 |
| Composto Meso em projecção de Fisher |
- Projecção de Fisher: util para compostos com vários esterocentros. Linhas verticais - para trás do plano. Linhas horizontais - para a frente do plano. Cadeia carbonatada de cima para baixo com todos os grupos eclipsados.
Compostos Ciclicos
- Ciclopentano: dois carbonos esterogénicos (os com CH3) → 4 isomeros → 2 meso → 3 formas numericas.
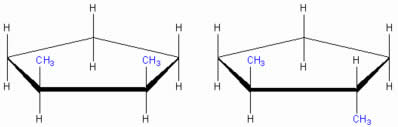 |
| Ciclopentano. Esquerda: composto meso (CH3 cis), aquiral, sobreponivel. Direita: forma enantiomeros (CH3 trans), imagem espelho. |
- Número de enantiomeros: 2^n (n - nº carbonos quirais).
- Derivados de ciclohexano
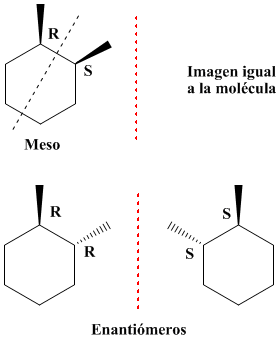 |
| Cis: plano de simetria. Trans: não tem plano de simetria. |
Relacionar esteroquimica do composto
- C esterogenico mantem-se, retenção da configuração.
- Alteração da prioridade, muda (R) e (S), há retenção da configuração.
Reacções iónicas: reacções de substituição nucleofilica e elminição de hidrogenetos de alquino.
Covalentes: partilha de dois electrões de igual forma pelos dois nucleos.
Reacções quimicas: é indispensavel a quebra e formação de ligações (E).
- Homólise: separação igual: A:B → A' + B'
- Heterolise: separação diferente. A:B → A+ (catião) + :B- anião
Reacções em vários passos: intermediarios.
Electrofilos: querem electrões para ficar mais estáveis.
 |
| Carbaniões vão tentar dar os seus electrõesl |
Reacção de substituição nucleofilica
- Substituição: entra um, sai outro. Nu:- + R-X → R-Nu + X-
(nucleofilo + halogeneto de aquilo → produto + ião halogeneto)
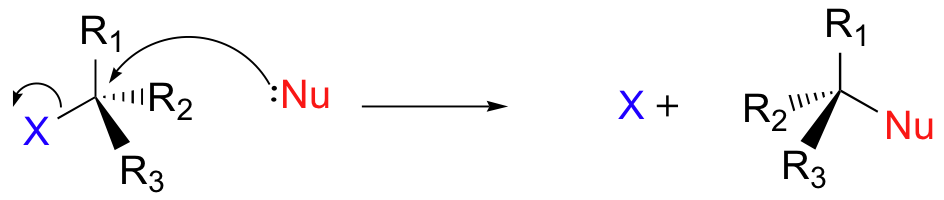
- Adição: só entra.

- Eliminação: só sai.
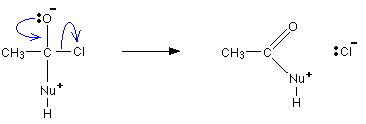
Leaving group: grupo que sai e que vai puxar electrões para si. Altamente electronegativo, pouco reactivo, estavel sozinho, carga net → base fraca.
 |
| Heterolise. O leaving group / halogenio tem grande electronegatividade (δ-) |
δ: relaciona-se com a densidade electronica e electronegatividade.
Velocidade de reacção - cinética
- Medimos em determinados intervalos a quantidade a que os reagentes se gastam ou a quantidade de produto que se forma.
- Teoria da colisão:
Vel. reacção = (nº total de colisões entre reagentes l/s) x (fracção de colisões com orientação correcta) x (fracções de colisões com energia suficiente para ocorrer reacção)
Substituição nucleofilica bimolecular (SN2)
- Tem estados de transição.
- Energia máxima por aproximação de um núcleo rompe a ligação existente.
- Estado Postular: determina a direcção da reacção.
- Reacção exotermica: ΔHº <0, expontanea.
- Energia de Activação: romper ligações, diferença entre a energia do estado de transição e a energia dos reagentes.
- Após formar a nova ligação liberta energia.
- Energia alta: reacção lenta. E baixa: reacção rapida.
- ΔH: explica a estabilidade dos compostos formados.
- > temperatura, > nº de colisões, mais rapida a reacção.
Esteroquimica de reacções SN2
- Velocidade depende de 2 substratos e não tem intermediarios reactivos.
- O grupo substituinte entra do lado oposto ao do living group, tem de haver mudança de configuração.
Reacção SN1
- Velocidade depende apenas do substrato e tem intermediarios rectivos (carbocatião).
- Envolve apenas um substrato no estado de transição.
- Realiza-se por vários passos.
- O estado de transição é o 1º passo e o determinante para a reacção, pois é o estado de transição o determinante da velocidade.
Estabilidade relativa do carbocatião
- Quantos mais grupos R estiverem ligados ao carbocatião, maior a sua estabilidade.
- Efeito indutivo (I): transmitido através da ligação, pode ser + ou -. Efeito dador de electrões (grupo alquilo).
Esteroquimcia de reacções SN1
- A molécula vira para o lado oposto da entrada do nucleofilo.
- Produtos (moleculas quirais) → 2 isomeros → provem da entrada pela frente ou por trás → forma racemica → 1:1 → opticamente inactivo.
Mecanismo: Brometo de alquilo terciario →1º passo lento → carbocatião estável (aquiral) → 2º passo rapido → alcool carbonado → 3º passo → perde protão → reacção acido base.
Comparar SN1 e SN2
Efeito da estrutura do substrato
- Mais reactivo: Metilo > Alquilo 1º > Alquilo 2º > Alquilo 3º :Menos reactivo.
Aumenta a dificuldade do nucleofilo dar densidade electronica ao carbono, diminui a velocidade relativa da reacção. - O mais importante é a estabilidade dos carbocatiões formados.
- Estado de transição → passo mais lento.
- Factores que estabilizam o carbocatião estabilizam o estado de transição.
- O nucleofilo não participa no estado de transição.
- Somento halogenetos de alquilo terciario reagem por mecanismo de SN1 porque os carbocatiões terciarios são mais estáveis.
- Postulado de Hammond
- R. endotermica: estado de transição mais parecido com produtos.
- R. exotermica: estado de tranisção mais parecido com reagentes.
- Efeito da concentração e Força do nucleofilo (só em SN2)
- Nucleofilo: procura o nucleo e dependendo da reactividade são:
- Forte: reage completamente com o substrato, reage rapido.
- Fraco: reage lentamente.
- O mesmo nucleofilo no mesmo atomo é mais forte quando tem carga do que quando não tem.
- As forças relativas dos nucleofilos relacionam-se:
- O nucleofilo com carga negativa é sempre mais forte que o seu acido conjugado.
- Quando o atomo nucleofilico é o mesmo, as nucleofilicidades são relativas à basicidade.
Mais forte: RO- (alcoxido) > HO- (hidroxido) > ião carboxilato > alcool > H2O - O nucleofilo dá electrões, o R empurra, é dador, por isso RO- é mais forte que HO-.
- Efeito de Solventes
- Próticos: 1 H ligado ao atomo altamente electronegativo.
- Apróticos: não há H ligados a atomos altamente electronegativos, ex. alcano, alceno.
- Ligações covalentes polares: normalmente originam compostos apolares.
- Reacções SN2
- Tiois (R-SH): nucleofilos mais fortes que o alcool (S > O).
- Solventes próticos: maior tamanho do atomo → > nucleofilicidade.
- Solventes apróticos: menor tamanho do atomo → > nucleofilicidade (ex. DMSO).
- Moleculas de solventes próticos: podem formar ligações de hidrogenio com o atomo nucleofilico de nucleofilos.
- Nucleofilo pequeno → carga concentrada → solvente cerca totalemnte → solvatação.
- Nucleofilo maior → prende nucleofilo.
- Reacção SN1
- Solvente polar prótico: estado de transição parecido com o carbocatião, estabiliza por solvatação → diminui energia de activação → aumenta a velocidade.
Natureza do grupo abandonador (leaving group)
- Melhor: base mais fraca, mais estável.
- I- > Br- > Cl- >> F-
- Efeito ressonacia → 3 estruturas de ressonancia:
- Electrões deslocalizados.
- Quantas mais temos maior deslocalização electronica, menor energia.
- Bases fracas: alquisulfato, alcanosulfato.
- Reacção de eliminação de Halogenetos de Alquilo (nada entra, tudo sai)
- Leva à formação de uma ligação multipla: de 2 σ → 1 π (C=C) e 1 σ (y-z).
- Unimolecular. no passo que controla a reacção só ha 1 molecula implicada - substrato.
- Bimolecular: no passo que controla a reacção existem 2 moleculas (base + substrato).
- Desidroalogenação: C-C + :B- → C=C + H:B + :X-
- Mecanismos de reacção E2: estado de transição envolve o halogeneto + base.
- Reacção E1: estado de transição envolve apenas o halogenato.
- Reacção de substituição nucleofilica biologica: metilação biologica
Metionina + ATP → S-adenosilmetionina (SAM) → fornece metilos (CH3)
Grupo trifosfato - bom leaving group, estabilizador por efeito de ressonancia.
Reacções de Radicais livres
- Muito reactivos, surgem por homolise.
- Energia de dissociação homolitica de ligação (DHº): energia necessaria para romper ligações covalentes homoliticamente.
Estabilidades relativas de radicais de alquilo
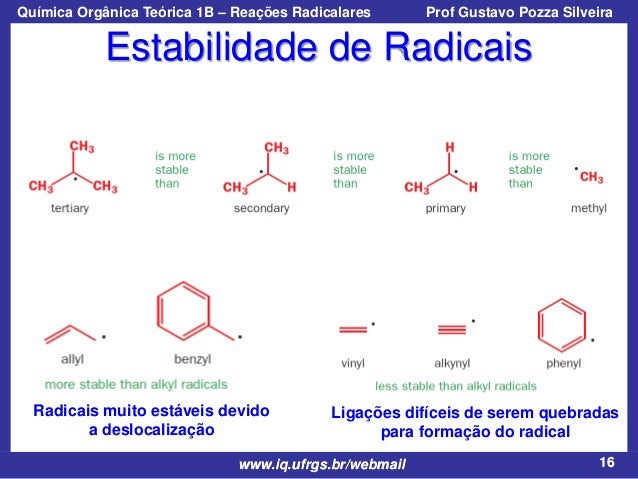 |
| Mais estavel por ΔHº menor, menor energia. |
Cloração de metano: mecanismo de reacção
- CH4 + Cl2 → CH3Cl + HCl + (CH2Cl2, CHCl3, CCl4)
- Clº ataca moleculas de metano sucessivamente → reacção muito extensa.
- Não continua indefinidamente - passo de terminação da cadeia.
Energia de activação
- Reacções com quebra de ligações, E act > 0.
- E act r. endotermicas >ΔH.
- E act = ΔH reacções gasosas onde só há quebra de ligações (homeoliticamente).
- E act de reacção de fase gasosa → combinação de radicais em moleculas → O.
Alcenos
- σ mais forte → alcanos têm σ → mais E para reagir.

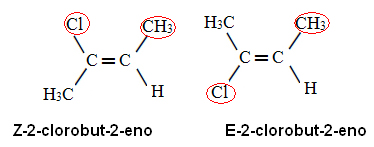 |
| Z e E. |
Propriedades fisicas dos alcenos e alcinos
- Apolares.
- Soluveis em solventes apolares (ou de baixa polaridade).
- Menos densidade que a água.
Hidrogenação de Alcenos
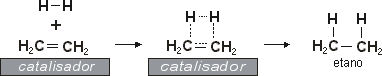
- Reacção exotermica.
- Reacção de adição: à temperatura ambiente com catalizador (Pt, Ni, Pd).
- Reacção em vário passos - fixa 2 moleculas ao metal depois aproxima-os para que se possam juntar → forma-se alcano antes de o libertar.
- Hidrogenação catalitica: adição syn, entram 2 H do mesmo lado.
- Hidrogenação sem catalizador: podem entrar de lados diferentes, anti.
- Formula geral:
- Alcanos: CnH2n+2
- Alcenos: CnH2n (deficiencia em hidrogenio).
Determinação da formula molecular
- Análise elementar: C, H, N, O.
- Espectroscopia de massa de alta resolução (HRMS).
- Hidrogenação permite distinguir ligações duplas, triplas ou em anel.
Estabilidade relativa dos Alcenos
- Cis-2-Buteno + H2 --Pt→ Butano ΔH = -28.6 kcal/mol
- Trans-2-Buteno + H2 - -Pt→ Butano ΔH = -27.6 kcal/mol
- Como E é libertada para formar o mesmo produto.
- É menor na trans, quer dizer que esta é mais estável.
Reacções de combustão
- Só podemos comparar o ΔH cem reacções com o mesmo produto final.
- O produto da reacção de combustão é sempre H2O ou CO2.
Estabilidade relativa de Alcenos
- Quanto maior o nº de grupos de alquilo ligados, maior a estabilidade do alceno.
Obtenção de alcenos para a desidratação de alcoois
- Facilidade: mais facil em alcool 3 (com 3 H substituidos por grupos).
- Mecanismo:
- Passo 1: necessita de ácido (rápido).
- Passo 2: formação de carbocatião (lento).
- Passo 3: formação de alceno (rápido).
- E act3 < E act2 < E act1
- Alcoois terciarios mais facilmente desidratados → carbocatiões terciários mais estaveis (energia de transição < E) → E act menor.
Estabilidade do carbocatião e rearranjos moleculares
- Alceno
- Dissubstituido: menos estável.
- Tetrassubstituido: mais estável.




0 comentários:
Enviar um comentário